转录组分析一般用于比较不同处理因素下,某组织或细胞的转录水平层面表达有何差异,并找到关键的差异基因。例如,比较某药物处理某肿瘤细胞后,与对照组相比哪些基因的表达水平有显著差异,这就是一个最简单的两独立样本差异表达分析。
转录组学研究的基本流程主要分为数据获取、数据分析两大步骤。
一、数据获取 #
转录组差异表达分析,是对两组样本的全转录组的相同基因进行对比,分析其表达量差异有无统计学意义。为了进行上述对比,需要获得两组样本全部基因的表达量,即表达矩阵。常规转录组测序生成的表达矩阵,如图1所示,是张二维表格,每列代表某个样本的全转录组,每行代表某个基因的表达量。

图1 表达矩阵示例
以图1为例,该表达矩阵是由 3 个实验组样本和 3 个对照组样本的基因表达量组成。样本可以是一批细胞,也可以是一块组织。
无论何种分析工具或软件,获取表达矩阵是一切分析的开始。表达矩阵由 RNA 测序数据生成。如果前期查询公共数据库,发现已有前人做过与自己课题类似的实验且提供了测序数据,可直接使用该研究的数据;否则只能自行设计实验进行测序。目前商业上的转录组测序已经十分成熟,提供样品测序之后,测序公司可以直接将表达矩阵返回给研究者,无需研究者用 FASTQ 等原始测序文件手动生成表达矩阵。
1. 查询公共数据库 #
GEO 数据库和 TCGA 数据库是转录组数据获取常用的两个数据库。
GEO 数据库是 NCBI 旗下的基因表达数据库,学术论文提交时若使用了测序数据,期刊往往会要求论文作者将测序数据提交至 GEO 数据库,所以 GEO 数据库的资源十分丰富。GEO 数据库中的测序数据不仅涵盖芯片、高通量、单细胞等转录组测序数据,还包含了非编码 RNA 等其他数据。

图2 GEO数据库
TCGA 数据库是 NCBI 旗下专注于肿瘤方向的基因数据库,涵盖 33 种癌症类型的 20000 多个原发性癌症样本及匹配的正常样本的基因组、表观基因组、转录组和蛋白质组数据,对肿瘤方面的的研究较为方便。

图3 TCGA数据库
2. 转录组测序 #
2.1. 芯片测序 #
基因芯片又称 DNA 微阵列(DNA Microarrays,吐槽一下, 有些翻译软件会将 array 翻译成 “数组”),原理是将已知序列的核酸片段固定于基板上作为探针序列,当待测样本中的核酸片段与探针序列原位杂交时,可以通过某些手段发出荧光从而被检测。通过芯片测序所生成的表达矩阵,每个基因的表达量通常是荧光信号强度的某种量化值。芯片测序表达矩阵的每一列中的每个基因,代表了该份细胞或组织样本中所有细胞对该基因的平均表达量。
芯片测序仅能探测到已设计在基板上的序列,无法探测到未知序列或基因。此外,由于基板上的探针序列存在饱和效应,因此无法探测表达水平过高的基因的真实丰度。芯片测序在高通量测序普及之后已较少使用,但 GEO 数据库中仍有数量众多的芯片数据可供利用。
2.2. 常规转录组高通量测序 #
常规转录组测序又称为批量转录组测序(Bulk RNA Sequencing,Bulk RNA-seq 或 RNA-seq),指对一份细胞或组织样本所有的 RNA 进行测序。目前测序公司可以直接将表达矩阵返回给研究者,无需研究者手动生成表达矩阵,以下内容仅作学习了解。
二代测序的结果是大量的短片段序列(reads 或 fragments),测序原理及相关概念请参考本站文章《核酸测序技术及原理》。要确定某个 read 属于哪个基因,需要将该 read 比对到参考基因组中。如果一些 reads 能唯一匹配到某个基因的序列,这些片段的数量(count)就代表该基因的表达水平。换而言之,Bulk RNA-seq 表达矩阵中,表达量代表的是唯一匹配到该基因的读段数(reads count 或 fragments count)。Bulk RNA-seq 表达矩阵的每一列中的每个基因,代表了该份细胞或组织样本中所有细胞对该基因的平均表达量。
将测序结果( .fastq 文件)匹配到参考基因组或参考转录组并生成表达矩阵,常用的工作流程有STAR-featureCounts、STAR-htseq 和 Bowtie-TopHat-htseq 流程。
2.3. 单细胞转录组测序 #
单细胞转录组测序(single cell RNA sequencing,scRNA-seq)是分别对一份细胞或组织样本中的每个细胞进行全转录组测序的技术。scRNA-seq 表达矩阵的每一列代表了样本中的一个细胞,每列中的每个基因,代表了该细胞对该基因的表达量。表达量代表的是唯一匹配到该基因的 reads count 或 fragments count。有关单细胞转录组研究的内容将在相关专题阐述,本文暂略。
2.4. 空间转录组测序 #
空间转录组测序(Spatial transcriptomics,ST-seq)类似于组织免疫组化或免疫荧光,能看到转录本表达量在组织学水平上空间分布的差异。没学过,有钱课题组的玩具,反正我这连 Bulk RNA 都拮据课题组是玩不起的( = = )。等学完了基础的分析再说。
二、数据分析 #
转录组数据或其他生信分析主要通过 R 或者在线分析工具实现。许多公司提供了简单易用的生物信息学在线分析网站,只要了解相关组学分析的基本原理及分析思路,不需要了解代码与编程,通过图形化的界面就能方便的分析数据,生成美观的可视化结果。然而现阶段的在线分析网站大多收费,或是分析的灵活度有所欠缺。
R 语言凭借开源免费多年积累的丰富生信分析 R 包生态,仍是生信分析的主流工具。本人笔记内容以 R 为主,阅读本人笔记的读者需要有一定的 R 编程基础。
1. 数据读入 #
表达矩阵常见的存储文件格式为.tsv或.csv或.txt(单细胞测序的表达矩阵除外)。这三者都是文本文件,主要区别在于,tsv 文件表格的各列以制表符(tab,”\t”)分隔,csv 文件以半角逗号分隔,txt 则无具体规定,可使用 tab、半角逗号、空格等分隔。在 R 中读取上述文件,可使用内置的 read.table() 、 read.delim() 、read.csv() 等函数。
2. 数据前处理 #
ID编码转换:不同来源的表达矩阵对于基因的编码往往不同。有的表达矩阵使用 Ensembl 数据库的 ID (Ensembl ID)编码基因,有的则使用 Gene Bank 的 Entrez ID。不同的工具包或数据库有时对基因的编码有具体的要求,可能需要进行编码转换。此外,为了后续分析输出的结果方便人的阅读,往往还需要将 Ensembl ID 或 Entrez ID 转换为人们更为熟知的基因名(Gene Symbol)。
基因过滤:原始表达矩阵中并未所有基因都有进行下一步分析的必要。例如有的基因在仅在个别样本中有表达且表达量极低,从生物学角度来看,一个基因必须达到某种最低表达水平,才有可能被翻译成蛋白质或在生物学上具有重要意义。此外,这些计数的显著离散性会干扰后续分析流程中使用的一些统计近似方法。因此,在进一步分析之前,应将这些基因过滤掉。不同的工具包有不同的过滤要求,建议参照工具的使用说明进行操作,例如单细胞分析流程中的数据过滤更加复杂。
重复基因合并:表达矩阵中,特别是芯片数据,许多探针探测的是同一个基因,以减少随机误差,因此会有很多行是同一个基因的表达量。在进入后续分析流程前需要将这些重复基因合并成一行。合并的方法需要结合工具使用说明或相关文献综合考量,有的分析工具建议选择仅保留表达量最高的一行;有的文献建议取所有行的平均表达量。
质量控制:样本的质量关乎到分析结果的可靠性。许多研究者会选择在差异表达分析前使用主成分分析(PCA)、箱型图等统计学手段,评估样本的质量、相关性等,以发现异常样本。
标准化(Normalization):标准化是通过某种数学变换,使两总体之间的某些参数在特定条件下可以互相比较的过程。例如,欲比较两个就诊患者年龄层构成不一致的医院对某病的治愈率,往往需要将两个医院各自的治愈率按年龄构成进行标准化之后再进行比较。
早期的差异表达分析会考虑对基因长度、GC比例、测序深度等因素进行标准化,出现了诸如每百万转录本数(Transcripts Per Million,TPM)、每百万 reads 数(Counts Per Million,CPM)、每千碱基每百万比对 reads 数(Reads Per Kilobase per Million mapped reads,RPKM)、每千碱基每百万比对片段数(Fragments Per Kilobase per Million mapped fragments,FPKM)等标准化方法。现在一般认为上述因素对于不同样本的同一个基因,影响是相同的,在仅对比同一个基因在不同组别的相对表达差异时无需标准化这些因素。
对差异表达分析有较大影响的因素主要是相对文库大小。高通量转录组测序中,某个基因的读段数(read count)会受到测序深度以及其他基因的相对表达丰度的影响。换而言之,RNA-seq 测量的是相对表达量而非绝对表达量。当某些样本中少数基因表达极高时,该高表达基因在特定样本中占据了总文库大小的很大比例,将导致该样本中其余基因的测序深度不足。若不对此效应进行调整,剩余基因在该样本中可能会被误判为表达下调。因此,需要标准化各样本之间的文库大小,最小化大多数基因在样本间的对数倍数变化。对于标准化的相关内容可以参考 edgeR 用户指南的 2.8 节。
3. 差异表达分析 #
3.1. 基本原理 #
差异表达分析是转录组学分析的一个核心步骤,目的是比较各处理组的样本之间,各个基因表达量差别是否具有统计学意义,即各组样本之间有哪些基因的表达水平是有显著差别的。
如图4,假设要比较 FN1 基因在实验组(蓝色框)和对照组(绿色框)之间的表达量差异有无统计学意义,这其实就是一个完全随机设计,两计量资料之间均数比较的统计学问题。

图4 差异表达分析示例
假设同组之间的基因表达量是正态分布,且各组之间方差齐,可以使用两独立样本 t 检验,P<0.05则差异有统计学意义,两组之间均数之比即为差异表达的倍数;对于多组方差齐的正态样本可以使用 ANOVA。实际上,read counts 只能取正整数,是离散变量而非连续变量,不服从正态分布,更近似服从负二项分布。负二项分布有其专用的分析方式,不宜使用上述正态分布的假设检验手段。
以 edgeR 包为例,其差异表达分析的统计学原理有一部分与 ANOVA 相似。在正态分布模型下,通常用标准差(方差开根)描述离散趋势,所以 ANOVA 使用离均差平方和(SS)将总体变异分解成组内变异和组间变异,即 SS总 = SS组内 + SS组间。在负二项分布模型下,离散参数的平方根是变异系数(coefficient variation,CV,标准差除以均数),描述二项分布模型下的离散程度。因此 edgeR 使用变异系数二次方根将总体变异分解成组内变异和组间变异,CV2总 = CV2组内 + CV2组间 。相关内容可以参考 edgeR 用户指南的 2.9 节。
3.2. 输出结果 #
芯片数据常用 limma 包进行差异表达分析,Bulk RNA-seq 常用 edgeR 包或 DESeq2 包。各工具包包内已经封装好了模型拟合的函数以及统计检验的函数,根据说明文档简单调用几个函数即可完成分析。图 5 为差异表达分析的结果示例。

图5 差异表达分析结果
图 5 所示的差异表达分析结果中,差异表达分析的结果一般使用 log FC 和 FDR 来描述。log FC 是差异倍数(Fold Change,FC)以 2 为底的对数值。FC 代表某组与被比较组相比(这里是实验组与对照组相比)基因表达水平差异的倍数,正值代表上调,负值代表下调。如图 ACTA2 的 log FC = 5.445,即实验组的 ACTA2 基因表达量是对照组的 43.562 倍。FDR 即统计学中的错误发现率(false discovery rate),又称为假阳性率、Ⅰ 类错误发生率,代表的是多重校验后矫正的 p 值。在多重校验中,随着检验次数的增加,假阳性率快速提高,此时需要使用检验的次数校正 p 值。有些分析工具的输出结果中,相对应的指标为矫正 p 值(adjust p value)。统计学上显著性水平一般取 0.05,即 FDR 或 adjust.p.value < 0.05 时差异有统计学意义。筛选出的差异基因可进行后续功能性分析。
实际科研中,为了可视化直观展示差异表达分析的结果,常绘制 log2 FC 对 – log10 FDR 散点图,将显著上下调的基因标上不同颜色,并标出最显著且差异倍数最大的若干个基因。这样的散点图形似火山喷发(大概吧),又称为 “火山图(Volcano Plot)”,如图 6 所示。
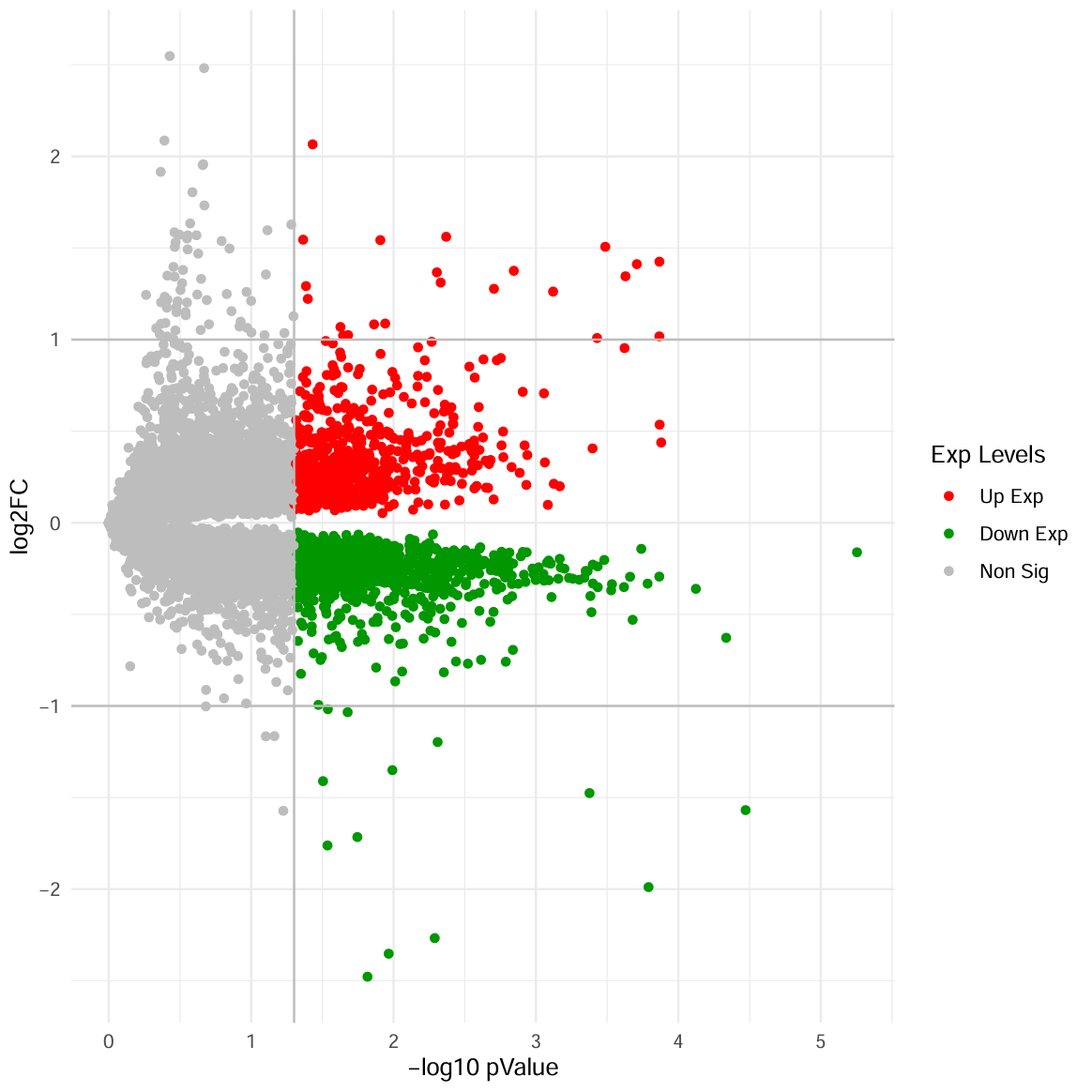
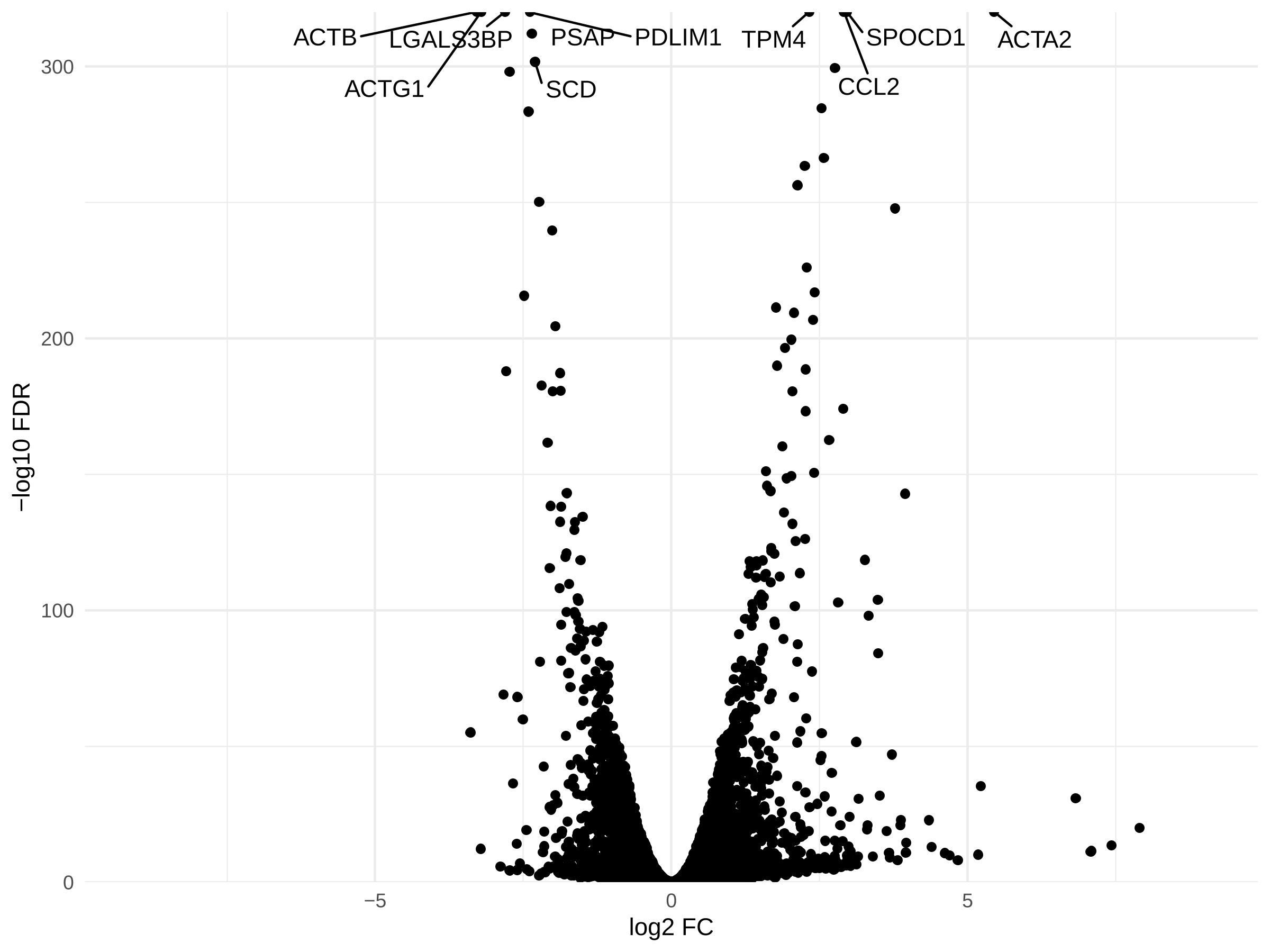
图 6 火山图
同组的不同样本之间由于处理方式相同,基因的表达模式趋于相似。将所有显著的差异表达基因按相似的表达模式聚类、按上下调标上不同的颜色、按表达水平高低标上不同的饱和度,绘制出各个样本的基因表达热图(Heatmap),这也是常用的数据可视化展示手段。如图 7 所示。
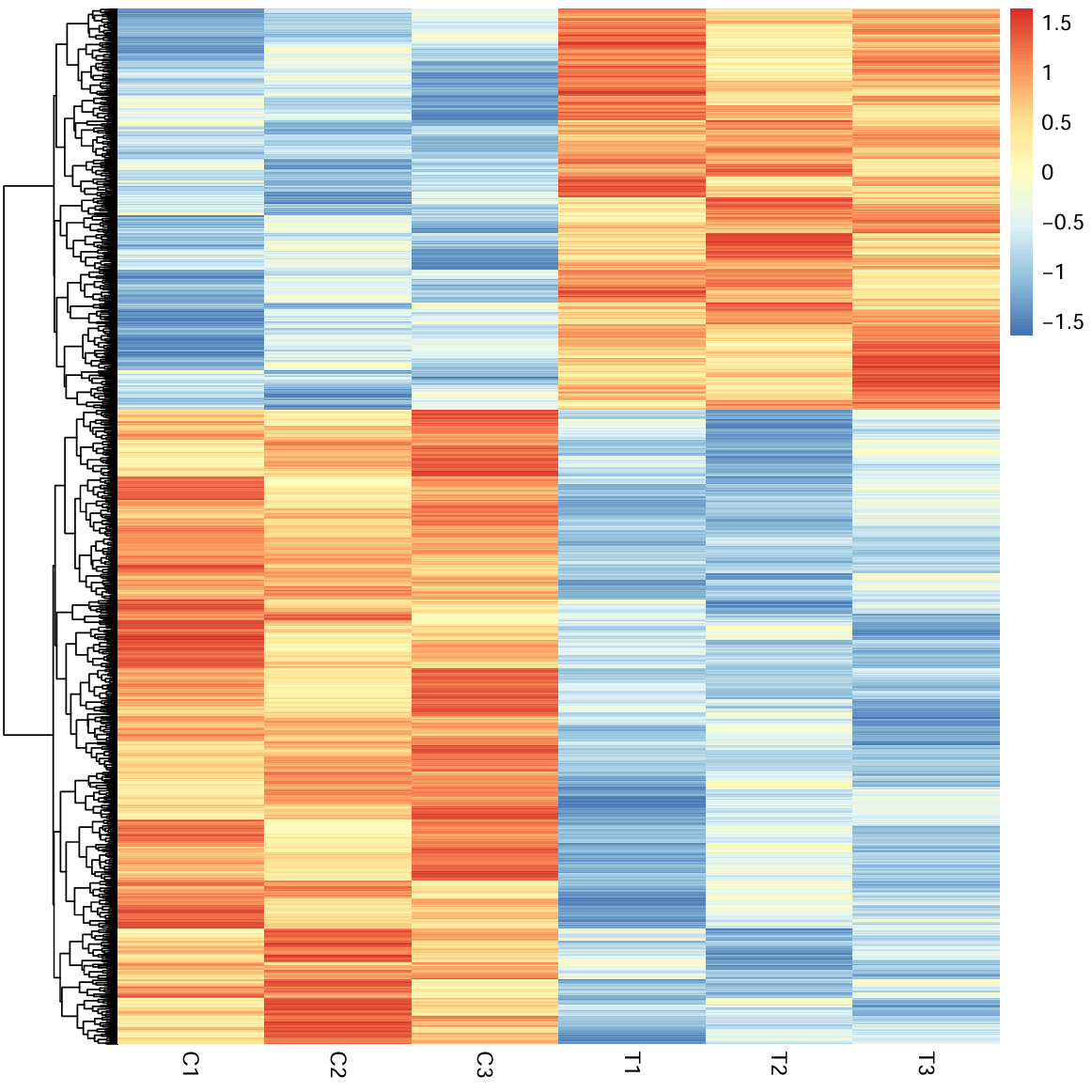
图 7 热图
图 7 横轴的 C1-C3 代表三个对照组样本,T1-T3 代表单个治疗组样本;纵轴中红色代表表达上调的基因,越红上调水平越高;相对应的蓝色则为下调。
4. 功能性分析 #
上一步的差异表达分析结束后,我们获得了两组样本之间的数千个差异基因。拿到这些基因之后如何进行下一步的课题研究呢?同时研究几千个基因显然是不太现实的,最自然而然的想法当然是,挑出几个 FC 最高、FDR 最小的基因使用 qPCR、WB、ELISA 等手段进行实验验证。
实际上,一个 mRNA 要翻译成蛋白质并实现其功能,还要经过翻译水平调控、翻译后水平调控,因此 mRNA 的丰度与其产生的蛋白质丰度或生物学效应之间并不是强相关的。单纯靠 FC 及 FDR 选择基因有失偏颇。其次,差异表达基因之间往往存在某种联系,可能从属于同一条信号通路,或是在代谢过程中具有协同作用。一开始就从单基因水平入手,往往会忽略基因之间的联系。
综上,获得一组差异基因之后,往往会进一步分析各基因之间的相互联系,其中常见的有富集分析(enrichment analysis)和共表达网络(co-expression network analysis)。
富集分析可理解为分析目标基因集共同集中于哪些信号或代谢通路、共同影响细胞何种功能。富集分析需要使用已知的基因功能注释信息作为先验知识。常用的基因注释信息数据库主要是 GO 数据库以及 KEGG 数据库。GO 数据库记录了每个基因在细胞中组成什么结构、发挥什么功能、参与什么细胞过程。KEGG 数据库专注于记录基因在什么代谢通路的哪个环节发挥什么调控作用。富集分析的内容会在相关专题中探讨。
基因富集分析依赖于功能注释的先验知识,不利于推测新的相互作用关系。共表达网络利用了相关功能的基因在特定环境中协调表达的特点,为研究基因之间的相互作用和调控关系提供了新的思路。共表达网络的内容会在相关专题中分析。
参考文献 #
[1]陈铭. 生物信息学[M]. 第四版. 科学出版社, 2022.
[2]周春燕,药立波. 生物化学与分子生物学[M]. 第9版. 人民卫生出版社, 2018.
[3]GEO Overview – GEO – NCBI[EB/OL]. Link.
[4]The Cancer Genome Atlas Program (TCGA) – NCI[EB/OL]. (2022-05-13). Link.
[5] Chen Y, McCarthy D, Baldoni P, et al. edgeR: differential analysis of sequence read count data User’s Guide[Z]. Link